2.2 Fusion gene Analysis
We used the FusionMap9 and our own in-house developed programs, FusionRNAscan, to predict candidate fusion transcripts from the RNA-Seq data. These two programs produce relatively small number of false positives since they require the fusion boundary to occur inside the sequence reads both in single and paired end data. Detailed procedure of prediction is as follows.
FusionMap prediction The RNA-Seq reads were aligned to the human reference genome (hg19) using BWA (version 0.5.9) with default parameters. All unaligned or discordant reads, which accounted for ~40% of total reads, were collected as an input file to FusionMap. The parameters were set as follows: the minimal end length of seed read (α) = 25, the maximal hits of read end (β) = 1, and the penalty of non-canonical splice pattern (G) = 4. In total, FusionMap reported 338 candidates of fusion transcripts, 132 of which were tumor- specific (114 normal tissue-specific candidates and 92 common candidates). We manually examined the BLAT alignments manually for tumor-specific candidates to remove reads whose head or tail part was mapped to multiple genomic loci. Among 55 uniquely mapped candidates, we selected 7 inter-chromosomal and 15 intra-chromosomal events as final candidates for experimental validation. Intra-chromosomal cases could be further divided into two groups, one with 5 events where fusion reads mapped onto opposite strands and the other with 10 events between non-neighboring genes on the same strand.
FusionRNAscan prediction Our in-house developed program is similar to FusionMap conceptually, but detailed logic and filtering procedures are substantially different. Most notably, we limit the candidates to those with genomic breakpoints inside the introns. Thus, the fusion boundary for both head and tail genes matches the respective exon boundaries exactly. Genomic alignment to hg19 was achieved by using the SSAHA210(version 2.5.5) program with ‘-solexa –skip 6’ option. Candidates for fusion transcripts should satisfy several stringent requirements: (i) minimum alignment length of 25 bp for both head and tail genes, (ii) the fusion boundary matching the exon boundary at exact position for both head and tail genes, (iii) fusion loci not in a repeat region or in a homologous region with the sequence identity of over 85%, (iv) consistent chromosomal identity in the non-fusion reads of paired- end data. Reads satisfying all of the above 4 conditions were named as seed reads. Setting the minimum seed read count as 2, we obtained 383 fusion candidates, 92 of which were tumor- specific. We further filtered out cases without valid gene symbols and cases with head and tail genes belonging to the same gene family. In an effort to find supporting evidences, we aligned the original RNA-Seq data onto the hypothetical fusion transcript to identify reads with partial overlap with either head or tail genes. These additional reads were designated as the rescued reads. After manual inspection of read alignments across the fusion boundary, we obtained 20 tumor-specific and 5 normal tissue-specific candidate fusion transcripts. Only tumor-specific cases were subjected to experimental validation as final candidates.
Experimental validation We carried out RT-PCR validation experiment followed by Sanger sequencing for 22 fusion candidates from FusionMap and 20 candidates from FusionRNAscan. In total, we confirmed 20 cases of genuine fusion transcripts (12 common predictions, 6 FusionRNAscan only, 2 FusionMap only). The list of confirmed fusion transcripts and primer pair sequences are shown in the Supplementary Tables 5 and 6. All candidates predicted by both programs turned out to be valid (12 cases). Candidates from FusionMap only were valid in 2 out of 10 cases. Candidates from FusionRNAscan only were valid in 6 out of 8 cases. Detailed examination revealed that two of those 6 confirmed cases were classified as multi-reads in FusionMap and were consequently eliminated at filtering steps.
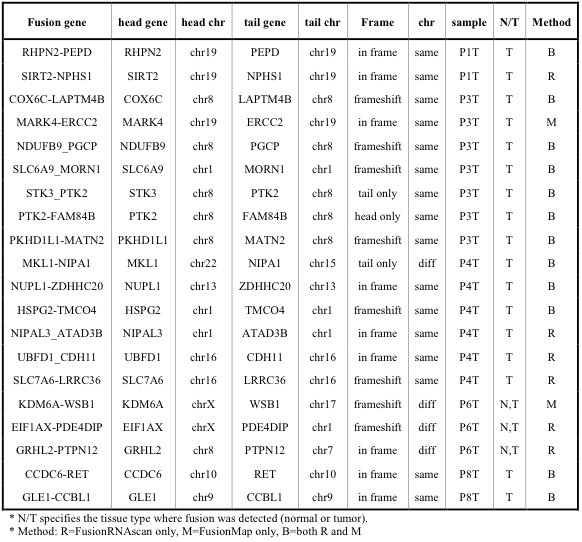
Supp. Table 5. List of fusion genes experimentally confirmed
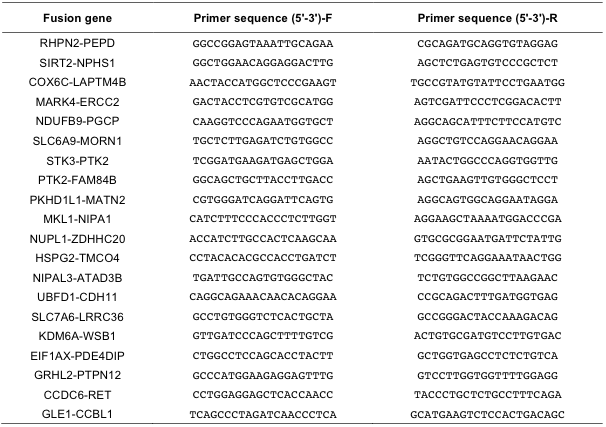
Supp. Table 6. Primer sequences for detecting fusion transcripts