1. Mutation Analysis
1.1 Mapping and mutation calling
Single-end 75bp reads from the exome-seq were mapped to the human reference genome (build 19) using the Burrows-Wheeler Aligner1 (BWA, v0.5.9). An average of 34 million reads was generated for each sample, of which 59% aligned to the targeted regions. Overall, 79% of the targeted regions were covered. Aligned reads were processed by Picard (http://picard.sourceforge.net, v1.42) to remove PCR duplicates. The Genome Analysis Toolkit2 (GATK, v1.0.5506) was then used to perform local realignment around indels and to recalibrate base quality score for producing a more accurate alignment for each sample. Supplementary Table 1 shows the statistical summary for mapping the exome-seq data for each patient.
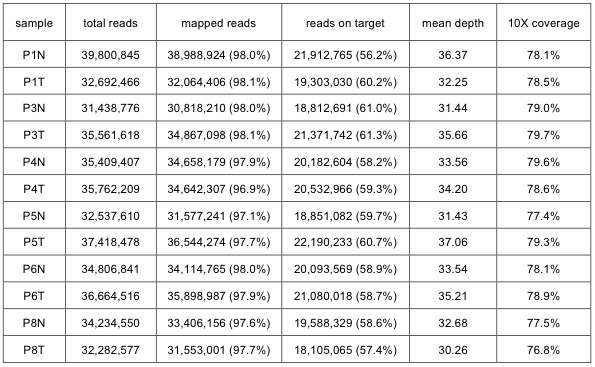
Supp. Table 1. Summary statistics for mapping exome-seq data
Mutation calling was achieved using the JointSNVmix3 (v0.6.2) to take advantage of the paired nature of the tumor and normal samples. Our initial attempt with VarScan4 (v1.0) produced numerous cases of what turn out to be false positives in subsequent validation experiments. Validation experiments were performed by PCR and conventional Sanger sequencing (with subcloning PCR products when necessary). JointSNVmix gives the posterior probability of somatic mutation (‘somatic probability’ in short) as the reliability index. Validation experiments showed that the somatic probability below 0.990 were mostly false. Using the cutoff values of 0.990 and 0.999, we obtained 335 and 189 candidates for somatic mutation, respectively. The GenomicAnnotator module in GATK and SIFT5 were used to annotate the variants in terms of coding potential and to predict the functional effects of the coding variants. The classification of somatic mutations with respect to gene structure and function is illustrated in the Supplementary Fig. 3.
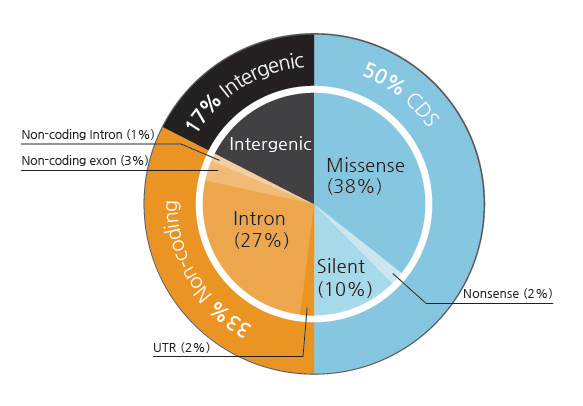
Supp. Fig. 3. Functional significance of somatic mutations
1.2 Experimental validation using Sanger sequencing
Among 189 candidates with somatic probability over 0.999, we selected mutations in CDS, UTR, and noncoding exons for experimental validation. Conventional Sanger sequencing following PCR amplification confirmed 45 valid mutations with 55 false predictions and 3 ambiguous cases out of total 103 cases. The prediction accuracy is below 44% even with the rigorous criterion of somatic probability over 0.999. Testing additional 4 candidates of probability between 0.990-0.999, we obtained just one positive out of 9 cases. Candidates of somatic probability below 0.990 turned out to be false in all 21 tested cases. We found one additional confirmed mutation predicted by VarScan only. This low success ratio may be due to the normal cell contamination and/or heterogeneity of cancer cells. In total, we identified 47 somatic mutations as listed in the Supplementary Table 2. The sequences of oligonucleotide primers used for PCR and sequencing are available upon request.
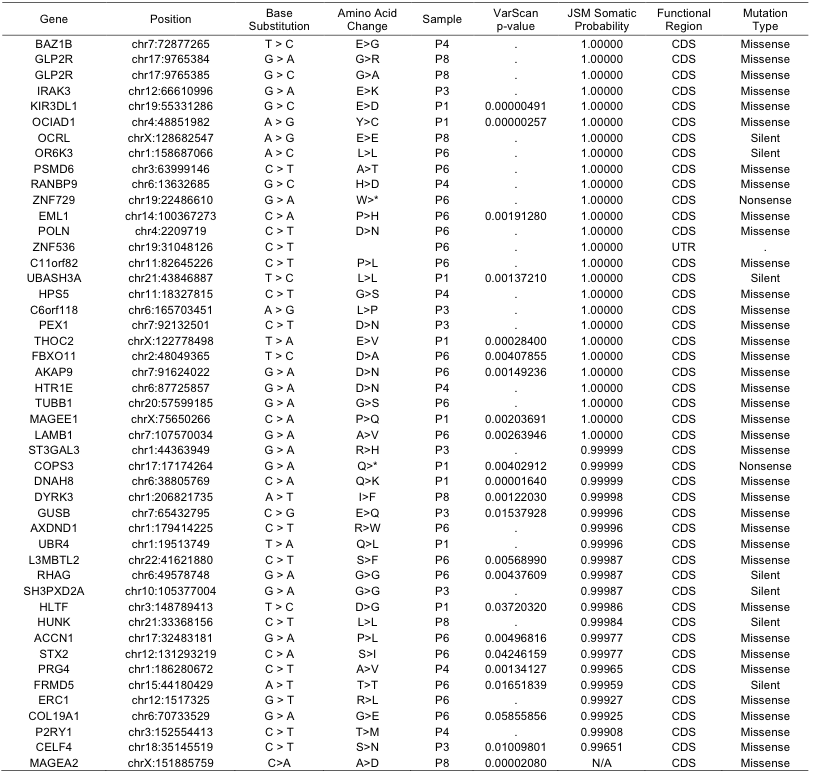
Supp. Table 2. List of somatic mutations experimentally confirmed